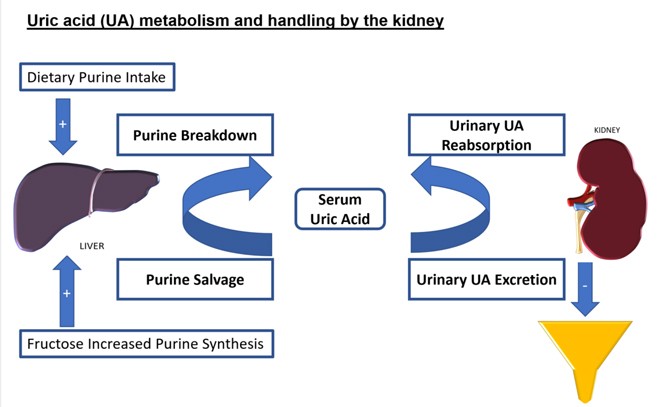
Product Candidate Pipeline
Our lead product candidates are XRx-008, XRx-101, and XRx-225. XRx-008 is in preparations for a Phase 3 registration clinical trial, the last stage of clinical development before application for FDA approval. Our XRx-101 program is advancing toward preparing for a “bridging” pharmacokinetic study for the Company’s Phase 3 clinical trial to potentially slow or reverse acute kidney disease in hospitalized individuals with respiratory virus infection. XRx-225 is at the non-clinical stage and advancing toward the clinical development stage.
Products
The Company’s most advanced development program, XRx-008, is a late clinical stage program focused on demonstrating the potential of our novel product candidate for ADPKD. XRx-008 is the development name given to XORTX’s proprietary oral formulation of oxypurinol, and shows increased oral bioavailability compared to oxypurinol alone. XORTX is also developing a second oral formulation of oxypurinol, XRx-101, for use in treating patients infected with respiratory virus infection with associated AKI.
XORTX is currently evaluating xanthine oxidase inhibitor candidates for the XRx-225 program to potentially treat T2DN as well as developing new chemical entities to address the large unmet medical need.
Patents
XORTX is the exclusive licensee of two U.S. granted patents with claims to the use of all uric acid lowering agents to treat insulin resistance or diabetic nephropathy, and two U.S. patent applications with similar claims for the treatment of metabolic syndrome, diabetes, and fatty liver disease. Counterparts for some of these patent applications have also been submitted in Europe. In both the US and Europe, XORTX owns composition of matter patent applications for unique proprietary formulations of xanthine oxidase inhibitors – U.S. and European patents have been granted. XORTX has also submitted two patent applications to cover the use of uric acid lowering agents for the treatment of the health consequences of respiratory virus infection. Recently, XORTX submitted a provisional patent application covering formulations and methods of treating individuals with renal insufficiency.
XORTX Therapeutics Pipeline:
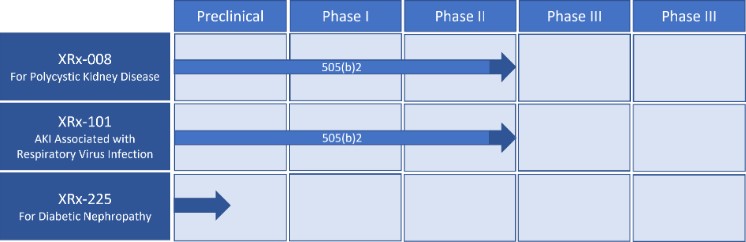
The interpretation by XORTX based upon FDA discussions is that the 505(b)(2) pathway and right of reference to the former NDA provide XORTX the ability to rely upon previous Phase 1 and Phase 2 clinical studies and/or conduct its own additional Phase 1 and Phase 2 studies for the XRx-008 and XRx-101 programs. However, we may elect to conduct our own Phase 1 and Phase 2 studies as necessary or required to gain marketing approval in the aforementioned programs. XORTX believes that the XRx-008 program will be eligible for accelerated review.
Our Strategy
Our goal is to apply our interdisciplinary expertise and pipeline-in-a-product strategy to further identify, develop and commercialize novel treatments in renal disease and indications related to health consequences associated with diabetes. To achieve this objective, we intend to pursue the following strategies:
| 1. | Subject to discussions with FDA, submit an NDA to the FDA, requesting review under the Accelerated Approval program, following the successful completion of the Phase 3 clinical registration trial of the XRx-008 product candidate program, which we believe could establish a new standard of care for ADPKD. |
53